Features
The aim of this project is to provide an easy to use toolkit for working with phylogenetic data. It is a library written in C++, which currently supports:
-
Placements. Working with evolutionary placements of short NGS reads on a
reference phylogenetic tree is the main focus of genesis. The data is typically produced by
tools like RAxML-EPA or
pplacer and usually stored in
jplace files.
- Read, manipulate and write
jplace files.
- Extract, filter and merge placements.
- Calculate distance measures (e.g., Earth Movers Distance).
- Visualize read abundances on the branches of the tree.
-
Trees. Reading, annotating and writing phylogenetic trees is supported, mainly for
visulization purposes.
- Read and write annotated Newick trees, write PhyloXML and Nexus trees.
- Versatile tree data structure that can store any data on the edges and nodes.
- Iterate trees with different policies (e.g., postorder, preorder).
-
Sequences. Some basic functionality for reading, filtering and writing sequences in
fasta and phylip format is available.
There are furthermore several detail functions, as well as a multitude of convenience and utility
functions and classes.
The feature list will hopefully grow in the future.
Getting Started
You find all the information for getting started with genesis in the
documentation.
It contains a user manual with setup instructions and tutorials, as well as the full API reference.
For user support, please see our
support forum.
It is intended for questions on how to do things with genesis.
For bug reports and feature requests, please
open an issue on our GitHub page.
Showcase
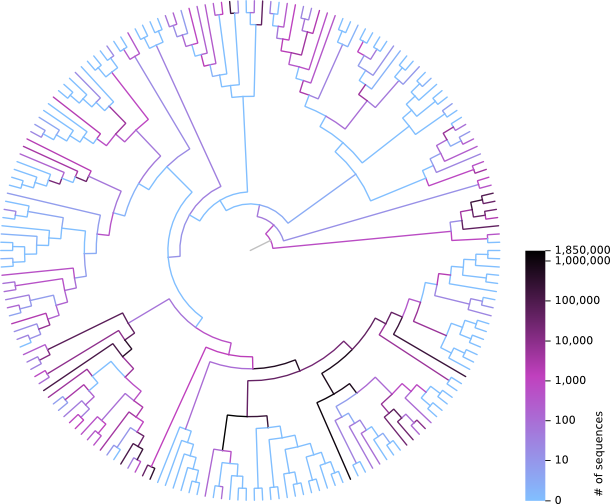
A focus point of the toolkit is to work with phylogenetic placements.
The following figure summarized the placement position of 7.5 mio short reads on a
reference tree with 190 taxa. The colour code indicates the number of reads placed
on each branch.
The color coding in form of a tree in Nexus format was produced with genesis by reading jplace
files of the placements,
and visualized using FigTree
and Inkscape.